Just over a week ago the long-awaited AlphaFold2 (AF2) method paper and associated code finally came out, putting to rest questions that I and many others raised about public disclosure of AF2. Already, the code is being pushed in all sorts of interesting ways, and three days ago the companion paper and database were published, where AF2 was applied to the human proteome and 20 other model organisms. All in all I am very happy with how DeepMind handled this. I reviewed the papers and had some chance to mull over the AF2 model architecture during the past couple of months (it was humorous to see people suggest that the open sourcing of AF2 was in response to RoseTTAFold—it was in fact DeepMind’s plan well before RoseTTAFold was preprinted.) In this post I will summarize my main takeaways about what makes AF2 interesting or surprising. This post is not a high-level summary of AF2—for that I suggest reading the main text of the paper, which is a well-written high-level summary, or this blog post by Carlos Outeiral. In fact, I suggest that you read the paper, including the supplementary information (SI), before reading this post, as I am going to assume familiarity with the model. My focus here is really on technical aspects of the architecture, with an eye toward generalizable lessons that can be applied to other molecular problems.
For AlphaFold2, the apparent answer that DeepMind gave to the question of what they should do is… yes. Self-supervision? Yes. Self-distillation? Yes. New loss function? Yes. 3D refinement? Yes. Recycling after refinement? Yes. Refinement after recycling? Yes. Templates? Yes. Full MSAs? Yes. Tied-weights? Yes. Non-tied weights? Yes. Attention over nodes? Yes. Attention over edges? Yes. Attention over coordinates? Yes. The answer, to all the questions, is yes! And this clearly paid off.
My somewhat flippant characterization may give the impression that AF2 is a mere smorgasbord of good ideas—this couldn’t be further from the truth. AF2 not only includes every conceivable feature but integrates these features in a remarkably unified and cohesive manner. It is as if all the team’s disparate ideas were repeatedly fed through the same intellectual bottleneck so that they emerge homogenized (their recycling approach, applied to the AF2 design process itself.) The result is both a tour de force of technical innovation and a beautifully designed learning machine, easily containing the equivalent of six or seven solid ML papers but somehow functioning as a single force of nature.
One sentence in the main text is particularly telling, as each phrase essentially corresponds to a whole paper, one of which has already been reported by a team at Facebook Research as a separate and impressive effort in its own right (the MSA Transformer; by all indications developed contemporaneously.)
“… we demonstrate a new architecture to jointly embed multiple sequence alignments (MSAs) and pairwise features, a new output representation and associated loss which enable accurate end-to-end structure prediction, a new equivariant attention architecture, use of intermediate losses to achieve iterative refinement of predictions, masked MSA loss to jointly train with structure, learning from unlabelled protein sequences using self-distillation, and self-estimates of accuracy.”
Suffice it to say that I am more impressed with AlphaFold2 after having read the paper than before. This is a work of art, one with numerous conceptual innovations that have nothing to do with compute power or “engineering” and is a more intricate tapestry than I had anticipated after the initial details were released. Soon after CASP14, I had a discussion with a friend in which the topic of how far the academic community was behind DeepMind came up. At the time I said it would have likely taken ten years of academic research to achieve what they did, while he thought it was more like two. After he said so I backtracked, thinking that perhaps I was being harsh. Now that I have read the paper, I think it would have likely taken at least 5-6 years before the academic community’s effort could have added up to AlphaFold2. (On this point, the right delta to measure is not RoseTTAFold vs. trRosetta, but trRosetta at CASP14 vs. Rosetta at CASP13; even then the gap is in large part due to the first AlphaFold.)
What I will do here is make a series of observations that summarize, from my perspective, the most interesting and surprising aspects of this unified architecture. They are (somewhat) listed in my perceived order of their importance.
Disclaimer: everything I say below is based on my understanding of AlphaFold2 and how it might be working, which may obviously be incorrect. It is a complex system with many moving parts, and I have no additional insights beyond what is provided in the Nature papers.
Table of Contents
- All roads lead to {si}
- Information flow is key
- Crops are all you need
- Always a refiner
- The why of SE(3)-equivariant reasoning
- The how of SE(3)-equivariant reasoning
- MSA Transformer
- DeepMind’s magic is not in brute forcing scale
All roads lead to {si}
The most important line in the whole paper, IMO, is line 10 of algorithm 20 in the SI, particularly when combined with the ablation of Invariant point attention (IPA) in Figure 4a in the main text, the core component of the structure module. If there is one takeaway for me, it is this line, as in it I think lies the crux of the “magic” of AF2. I say this not because of the line itself per se, but because of what it implies.
Let me back up a minute.
One of the challenges of building ML systems that reason over proteins is the fact that proteins are long 1D polymers that fold in 3D space. Key here are “1D”, “3D”, and “long”. On the one hand, given the sequential nature of proteins, it is natural to encode them using sequence-based architectures. When I developed the RGN model I did just that, using what was at the time the leading architecture for sequence problems (LSTMs). However, architectures of this sort have trouble reasoning over long-range interactions, a common phenomenon in proteins. Still, 1D representations are convenient because they readily map to the physical object, permitting parameterizations (e.g., internal coordinates) for predicting protein structure in a straight-forwardly self-consistent manner.
Most of the field, up to and including the first AlphaFold, pursued a different approach that relies on 2D matrices to encode structure using inter-residue distances. This captured long-range interactions better (relative to sequential models) but introduced the awkwardness of mapping a 2D representation to an object that is fundamentally a 1D curve in 3D space. Typically this mapping was accomplished using physics-based relaxation or SGD (the first AlphaFold) in a post-processing step, but left much to be desired.
When DeepMind first revealed AF2 at CASP14, they described what appeared to be a hybrid 2D/3D approach, in which the structure is initially encoded in a 2D representation that is then transformed into a 3D representation. There was much speculation about how this was done (more on this later), but it left, in mind, this awkwardness about the 1D-2D mismatch. This is a subtle point. The question here is not about how to map a 2D distance matrix to a 3D set of coordinates (many approaches exist.) Instead, it is about the structural mismatch (in terms of data / tensor types, not protein structure) between an object that is fundamentally one-dimensional (the protein itself) and its distributed representation in a 2D matrix. One can come up with various hack in terms of averaging rows and columns to transform the 2D representation into a 1D one, but they always struck me as just that, hacks.
What I did not anticipate, and what I find quite elegant about the AF2 architecture, is the central role that the MSA representation, and in particular the first row of that representation—which initially encodes the raw input sequence but is ultimately projected onto an object they denote 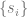
This was the most mind-blowing aspect of the paper to me, the part that felt most “magical”. And the way they’ve constructed it is very clever. It is indeed the case that learning using a 1D representation is difficult, especially for long-range interactions. So what AF2 does is use a 1D object to represent the protein, but couple it to a 2D object (the pairwise 
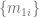
While I was reading the initial manuscript and making my way to algorithm 20, I felt rapt in anticipation of how the structure will finally be extracted. When I discovered that it was
In a way what the AF2 team has done, if I am allowed to speculate here a little, is to develop an approach where the representation used for learning and reasoning is decoupled from the representation used to hold the state of the system and predict structure, yielding the best of both worlds. In most ML architectures, a single representation is used to do both; in AF2, it is split. When DeepMind first announced their hybrid pair/MSA representation, I didn’t really get the “point” of it, but now that I understand it in detail, I suspect the above was one of their key motivations, although of course I have no way of knowing for sure.
Side note: when they incorporate structural templates, they add torsion angles as rows to
Information flow is key
If there is one unifying theme to the paper, one broad principle, it is that AF2 is engineered to maximize information flow between its components (all greased by liberal but intentional use of 
I will illustrate this with a couple of concrete examples. First is the communication between the pair representation 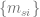


Another example is the communication within the pair representation, which utilizes two forms of novel “triangular” attention performed between pairs of pairs of residues—naively a very expensive computational operation. Here too efficiency is achieved by using a light touch approach of only biasing the attention, and more importantly by restricting attention to pairs of pairs that have one residue in common, with the intuition being that the triangular inequality ought to constrain such quantities (because three residues form a triangle.) This one is interesting because it illustrates an important principle: geometric constraints need not be realized literally, as many people, including my group, had been trying to do, by e.g., mathematically enforcing the triangular inequality, but instead informationally, i.e., in the information flow patterns of the attention mechanism. In effect, what AF2 does is convert geometric intuition—we know these two pairs of distances ought to constrain one another—into architectural features that allow these pairs of distances (or rather the representations encoding them) to communicate easily. This to me is a general principle of ML architectural engineering, particularly for problems with rich domain knowledge. It illustrates how prior information ought to be integrated into learnable models: not through literal hard constraints or even soft ones, but as aspects of the learning process itself. (I would say this is broadly true of deep learning, vis-à-vis say probabilistic programming or Bayesian modeling. Knowledge is rarely injected directly in DL; it is instead infused into the architecture through designs that make it easier to learn. In this sense, hard SE(3)-equivariance is actually an exception, but more on that later.)
One last example is in the IPA structure module, which when enabled gets both a light touch from the pair representation in line 7 of algorithm 22, where
Crops are all you need
AF2 (and the first AF before it, although not quite in the same way or extent) is trained in a seemingly strange way: not on entire proteins, but on fragments of ones, or what the AF2 team calls ‘crops’. They are not short; usually a couple of hundred residues. But for longer proteins, these crops only capture a small fraction of the whole sequence. Furthermore, two non-contiguous crops are often made (from the same protein), and then stitched together. Remarkably, while AF2 is mostly trained on crops of up to 256 residues (later fine-tuned on 384), it can predict protein structures with well over 2,000 residues, an astonishing feat. Not only because it is a very hard problem in absolute terms, but also because AF2 is trained on much shorter crops.
Global context matters in proteins. The same subsequence of amino acids, in two otherwise different proteins, will not in general have the same structure; protein structure prediction would have otherwise been solved long ago! (Of course, this gets progressively less true as the length of the subsequence grows.) Taking disembodied crops, that can be hundreds of residues apart, and asking the model to predict their relative orientation when even the length of their separation is unknown (if they are more than 32 residues apart) seems like an impossible task. But there are at least two ameliorating factors at work here. First, AF2 is working with MSAs / co-evolution patterns, which encode this information irrespective of linear chain separation. Second, this is only done at training and not inference time. During inference, AF2 does have access to the whole sequence, and so the issue of context-sensitivity is moot. During training, AF2 may get one signal from one pair of crops in one protein, and a conflicting signal from a similar pair of crops in a different protein, and that would be totally fine. Such situations likely teach the model that there is inherent uncertainty when it sees these two crops at some unknown separation. It is being taught the context-sensitivity of proteins. Recall that, during training, there is no requirement that the model is ever able to accurately predict the structure. All that matters is that useful information is being imparted to the model through gradient updates.
This is another instance of decoupling usually coupled things (the previous one being suitable representations for learning and holding state.) In most ML models, the training and inference tasks are kept very similar, with the idea that the more similar the training task is to the inference one, the better. But this is not really necessary, given that training is about acquiring useful signal from the data while inference is about making accurate predictions. This idea is certainly not unique to AF2; generative models often involve quite different tasks during generation vs. training, and of course differentiable loss functions used during training are often different from the true target functions. Still, AF2 demonstrates a rather robust use of this idea in the supervised learning context, almost arguing for intentionally decoupling tasks, when in general people have tended to treat such decouplings as failures of modeling.
In fact, it appears that the architects of AF2 had this in mind as a design feature, an inductive prior for the model. This shows up most visibly in the relative positional encoding of residue pairs, which are capped at 32 residues apart (i.e., in terms of raw inputs, if two positions are more than 32 residues apart, they are treated as 32 residues apart.) For contiguous crops, this is not really a limitation because the model can learn to add positional embeddings so that it can infer the true separation. But for non-contiguous crops, it will have no idea what the separation distance is. Yet, the model is being tasked with figuring out how the two crops should be oriented with respect to one another. This is an inductive prior that states that, in protein-land, if two crops are beyond a certain distance apart, it doesn’t really matter how far apart they are exactly. It is an interesting prior, and one that seems to have paid off for AF2. [Update 7/27/21: Turns out that the multiple non-contiguous crops scheme was a feature of the manuscript I reviewed but not the final version, as it was not used in the CASP14 model of AF2.]
I don’t know if the above was the intention for the AF2 team going in—memory efficiency likely played a big role in their thinking, given the near impossibility of training something as large as AF2 on full-length proteins using current TPUs. Nonetheless, what may have started out as a computational trick ended up being a good idea biophysically-speaking.
Incidentally, this resolves one of the great mysteries about AF2 during CASP14—the gap between inference and training times. Because of cubic scaling, and the fact that inference is done on full-length proteins while training is done on crops, there (can be) a very large compute-time gap between the two.
Always a refiner
Another striking feature of AF2 is its always-on refinement mode. I.e., it is always capable of taking a preliminary structure at some distance from the native state and refining it to be closer to that native state. This is true in multiple modules and at multiple levels of granularity, making the system remarkably robust and versatile at utilizing diverse types of data.
It is most obvious and natural in the structure module, where the weights of the iterative IPA procedure are tied and so the same operations are applied repeatedly. This makes sense as IPA’s intended function is to refine the structure coming out of the evoformer. However, the evoformer itself is also always in refinement mode. This is not explicitly encoded in the architecture per se (the weights of the 48 layers of the evoformer are untied) but is evident in the way it is trained, where it is encouraged to behave this way. For example, the raw inputs can include templates of homologous structures, some of which may be similar to the sought structure, thus providing the very first layer of the evoformer a structure (encoded in the pair representation) that is essentially complete and that should not be screwed up. This is key and encapsulates what is hard about this, because AF2 may also get a sequence that has no structural homologs, thus providing the first layer of the evoformer with virtually no structural data—in both instances, the evoformer must learn to behave correctly. Repeated subsampling of the MSAs reinforces this, because each sample provides varying degrees of sequence coverage.
The same phenomenon also occurs with recycling. First, recycling itself is a form of refinement, as the entire network, with tied weights, is reapplied up to three additional times. But the act of recycling also teaches the evoformer to be a refiner, because the same evoformer in a later recycling iteration can be presented with structures that are much further along than the evoformer in the first (pre-recycling) iteration.
Another mechanism to encourage refinement is the use of intermediate losses, both in the structure module and in recycling. I.e., during training the model is optimized to minimize losses of the final predicted structure as well as those of intermediate structures predicted some fraction of the way through the system. This encourages AF2 to not only predict structures correctly, but to do so quickly, in earlier iterations of the system. In the structure module this is done very explicitly; its loss function is literally an average over all iterations. In recycling it is a bit more subtle. The loss from only one iteration is used for backpropagation, but because the number of iterations is stochastically sampled, the effect is the same; the model is encouraged to get structures right in earlier recycling iterations.
AF2’s robustness to varying tasks at varying stages is evident in the animations supplied with the paper. The video of LmrP (T1024) shows a structure that is essentially complete after the very first layer of the evoformer, while that of Orf8 (T1064) goes on and on till the very end, almost resembling a molecular dynamics folding simulation (it is not one, obviously.) Incidentally, these animations are also suggestive of AF2’s behavior with respect to sequences of varying MSA depths. For deep MSAs, it perhaps acts similarly to pre-AF2 methods that relied heavily on co-evolutionary signal, inferring the structure more or less entirely based on that signal with just a single evoformer layer. For sequences with very shallow MSAs, it falls to the later stages of the evoformer and structure modules to actually fold the protein, where I suspect the model is learning and applying general physics knowledge about protein structure. The “No IPA and no recycling” panel of Supplementary Figure 5, which shows AF2’s performance degrading substantially for shallow MSAs when recycling and IPA are turned off, supports this hypothesis. Furthermore, AF2’s apparent success in predicting proteins complexes from unpaired MSAs may be due to this general physics knowledge, although Sergey Ovchinnikov has a compelling alternate theory.
On the whole I find the idea of constant refinement powerful and broadly useful, especially when it can be applied without having to backpropagate through the entire model. I am not sure if this is novel within the iterative supervised learning context (it probably has parallels in generative computer vision). It is very RL-like, which is obviously DeepMind’s forte.
The why of SE(3)-equivariant reasoning
Possibly my biggest surprise reading the paper occurred when I came across the ablation studies of Figure 4a, particularly the ablation of IPA, AF2’s much ballyhooed SE(3)-equivariant Transformer. While I did not ascribe as much value to this module as I think others did pre-publication, the fact that removing it seemed to do so little was still shocking. What was the point of all this work and machinery if it contributed so little?
There were actually two surprises here. First is the fact that without IPA, AF2 simply spits out 3D coordinates, without any explicitly SE(3)-invariant conversion of the “distances” in the pair representation to 3D space. In fact, as I mentioned earlier, the IPA-less version of AF2 relies entirely on the 1D
The second surprise is the fact that reasoning in 3D, i.e., reasoning after an initial version of the structure is materialized in a global reference frame, appears to not be terribly important, unless recycling is also removed. This flies in the face of our intuition that certain spatial patterns, particularly ones distributed across multiple discontiguous elements of the protein, are more readily apparent in 3D space, and should therefore benefit from 3D reasoning. From a practical standpoint, it also seems to obviate all the methodological research that has gone into equivariant networks, at least insofar as it applied to proteins (more on this in the next section.)
This is certainly one interpretation, but I don’t consider it to be an entirely accurate one. The key lies in the fact that removing IPA is ok only as long as recycling is retained. When both are ablated, as Figure 4a shows, performance drops considerably. Furthermore, if recycling is removed but IPA is retained, then AF2’s performance stays nearly unperturbed. This is a rather impressive showing for IPA, given that recycling is essentially quadrupling the 48-layer evoformer (computation cost-wise it is not, because of various tricks), while IPA is only 8 weight-tied layers. Viewed in this light, IPA layers are far more efficient than evoformer layers, at least for late-stage refinement. As the focus now shifts from single chain prediction to complexes and higher-order assemblies, the importance of spatial reasoning will only increase, and I expect that IPA and its future derivatives will continue to play an important role.
The how of SE(3)-equivariant reasoning
Setting aside the utility of SE(3)-equivariance, the question of how it is performed in AF2 was probably the most anticipated one prior to the publication of the paper. It helps to step back for a moment and consider where this subfield has been going for the last few years. The flurry of recent activity in equivariant neural networks arguably got started with a paper from Thomas et al., although there were some antecedent works. The paper relied on group-theoretic machinery, employing (convolutional) filters that used spherical harmonics as their basis set. The formulation is mathematically elegant and has been elaborated in numerous papers since, with one in particular by Fuchs et al., the SE(3)-equivariant Transformer, that not only generalized the approach from convolutions to self-attention but also shared an identical name to that used by the DeepMind team during CASP14 to describe what they now call IPA. This naturally led to the speculation that AF2 used something very similar to this approach, including in my own previous post on AF2. This, in retrospect, had little merit, especially since the approaches were developed around the same time and so there was no reason to believe they influenced each other.
Parallel to the development of group-theoretic approaches, there has also been a flurry of graph-theoretic approaches to the problem of reasoning equivariantly over molecules. Instead of relying on spherical harmonics, the graph-based approaches embed molecules as graphs, with spatial information encoded in the edges that connect nodes that in turn encode atoms. This line of research has been applied both to small molecules and to proteins, but arguably it is in the latter where it has found the most utility. Proteins, like all polymers, inherently permit the construction of unambiguous reference frames at each atom, and this fact has been exploited to great effect by graph-based approaches. One of the first—perhaps the very first—method to use this construction in the context of machine learning and proteins is the Structured Transformer by Ingraham et al., and it appears that this work was the inspiration for IPA. There are many advantages to using graph-based constructions over group-based ones for proteins, but this is a longer discussion that does not impinge on how IPA works. Suffice it to say that IPA falls squarely in the graph-based camp, and for proteins this IMO makes the most sense.
How IPA works is quite interesting, as it is arguably the most novel neural primitive in the whole of AF2, combining multiple spatial reasoning mechanisms that will likely inform much of future molecular modeling. I will focus on a few. I should emphasize that most of my thoughts are highly speculative, especially the ones on meta-reasoning.
First is the IPA attention mechanism, which is something of a beast (line 7 in algorithm 22). It includes the usual non-geometric query/key matching (

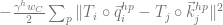

Second is how values, specifically geometric values in the form of 3D vectors, are communicated between residues (lines 3 and 10 of algorithm 22.) Each residue again sprays multiple vectors, all transformed to the global reference frame, where for any receiving residue, the vectors of all other residues are averaged in Euclidean space (weighted by attention and done per head/point value) over the whole protein before they are transformed back to the local reference frame of the receiving residue. I imagine this enables sophisticated geometric reasoning capabilities, ones that may reflect general aspects of protein biophysics and are less tied to the genetic / co-evolutionary information contained in MSAs.
Consider for instance a “catalytic triad” in which one residue must detect how two other residues are positioned and oriented with respect to itself. It sprays query vectors with the appropriate positions and orientations, and because they are specified in its local reference frame, they form a generic spatial pattern that IPA can learn and apply repeatedly. If the query vectors are pointing in the right direction, we can imagine key vectors that simply state the location and orientation of each residue, essentially returning 
Whether the above is an accurate description of how IPA works is of course unknown, and may not be knowable unless one carefully inspects the behavior of the learned weights and model.
Third, and by far most speculatively, is the possibility that IPA may combine its components to perform meta-reasoning. By this I mean reasoning not about protein structure, but about AF2 itself, namely the status of its knowledge about the current inference task and how it might be improved in subsequent IPA iterations (I am referring to learning in real-time during inference, not through gradient descent.) Imagine that AF2 expects one protein fragment to interact with another but is uncertain of its location. During the first iteration, it sprays multiple broadly spaced query vectors, distributed over a large section of the protein region that it thinks may contain the sought fragment. Nearly all queries won’t find a match, but one of them, if AF2’s hypothesis is correct, might. Once found and based on the information obtained, AF2 can in subsequent IPA iterations send more directed queries to better localize the exact position and orientation of the fragment, and then refine the structure according to its learned knowledge of proteins.
The key point here is the use of the iterative aspects of IPA to control its reasoning and discovery process. Compared to traditional sampling approaches, where different protein conformations are randomly considered, IPA (and the evoformer) may actively reason about how to improve its knowledge about the protein it is currently trying to fold. To be sure, there is no guarantee that this sort of meta-reasoning is happening, and I suspect it would be non-trivial even for DeepMind to assess this. If meta-reasoning is happening, it may explain the long folding times for Orf8 and other structures with shallow MSAs (in the sense of arriving at the native state.) If AF2 sprays space with vectors to find and orient protein fragments, it would be doing a form of search, one that can take many iterations to conclude. This would explain why the same computation, repeatedly applied, can eventually fold the protein, and the specific importance of IPA and recycling to proteins with shallow MSAs in the ablation studies of Supplementary Figure 10.
One last point: it is remarkable to me how, again, in line 10 of algorithm 22, all this geometric information is encoded back in
MSA Transformer
This is a minor point methodologically but an important one performance-wise. AF2 includes the MSA Transformer as one of its auxiliary losses, and Supplementary Figure 10 shows it to be critical for sequences with shallow MSAs, where performance degrades substantially without it. To me, one of the most impressive aspects of AF2 is its robustness to shallow MSAs. Self-supervised learning over sequences (or MSAs in this case) has long seemed like a natural way to tackle the problem, and it pans out here.
Interestingly, this gain evaporates when the MSA is very shallow, e.g., just a single sequence, but this makes sense too because then the MSA Transformer is uninformative.
DeepMind’s magic is not in brute forcing scale
I will end with perhaps the most counterintuitive conclusion I took from the paper. Going in, I had anticipated that at least for some components, most notably the evoformer attention mechanisms which, without simplifications like axial attention, would scale rather horribly, DeepMind had exercised their enormous computing resources to brute-force their way through problems that other groups would have to be clever about. The irony is that the truth appears to be almost exactly the opposite. What is impressive about the AF2 effort is not top-notch hardware, but top-notch software and ML engineering that renders brute-force scaling unnecessary. This part I suspect will be hardest for academia to replicate, because it is less about increased national investment in computing resources (which would undoubtedly help) and more about in-house professional software engineering capacity, a much taller order.
This is evident in numerous places: the careful use of gradient stopping, in IPA but also in recycling, where an enormous amount of compute is saved; in cropping, probably the most important and impressive, because it so fundamentally changes the inductive bias of the system; and in thoughtfully chosen initialization schemes. It is true of course that DeepMind can have their cake and eat it too. Their use of 8 ensembles for CASP14 demonstrates this, where they increased compute requirements by an order-of-magnitude for what appears to be very marginal gains (I suspect they only discovered this after CASP14 however.) But on the whole, their culture appears to be one of computational frugality, in the best sense of the word, and quite possibly the biggest compliment I can give the AF2 team on an achievement that is remarkable in so many other ways.

Hi – great blog as usual! I’m a bit puzzled about the bit describing the discontinuous crops that are stitched together. I’m struggling to see where that’s described in the paper. Is it something you’ve found in the code or am I just missing the key paragraph in the paper’s supplementary? I know in the first AlphaFold they used off diagonal crops, but I really must have missed where that’s described in the new one.
Thanks for bringing this to my attention. It was described in the earlier versions of the manuscript but removed in the final version, as it was not ultimately used in the CASP14 version of AF2. I have updated the text to reflect this fact.
Ha ha – how interesting! Thanks for the clarification.
Having said that, I do wonder if important undocumented changes like that have now crept into the released models – or perhaps the models being used to generate the EBI hosted data. It would be good if DeepMind could produce a change tracking document explaining how the current models differ from the original paper description. This is vital now the models are being distributed to biologists as finished products to be used in further experimental work. Normally I’d say check the code to be sure, but I don’t believe DM have released their training code, so we can’t.
I think this will likely to be a running concern–they say on the EBI website that the structures will be updated as newer versions of AF are developed, and I’m not sure how frequently they will be publishing on new variants of the model.
Interesting… Why not negative dot product and instead negative squared distance between q and k?
Correction — positive dot product
Hi I love your post but I have a small question. I looked up the Figure4(a) in the original paper and also figure10 in supp info, but I didn’t get why IPA is more ‘important’ than recycling cause no IPA do little on the result , but without recycling the LDDT drop a lot. I kind of feel confused about it and much appreciate if you share your insights
Hi I love your post but I have a small question. I looked up the Figure4(a) in the original paper and also figure10 in supp info, but I didn’t get why IPA is more ‘important’ than recycling cause no IPA do little on the result , but without recycling the LDDT drop a lot. I kind of feel confused about it and much appreciate if you share your insights
Pingback: The application of AI in Protein Structure Prediction – Cheat Sheets for Computational Biochemistry
Hi, I’m just trying to wirte a bachelor thesis on protein structure prediction and AlphaFold so I don’t not even have 1% knowledge of yours 🙂
But it seems to me there is an error: it gets confused column-wise with row-wise, in “However, the fact that {z_ij} affects the column-wise attention of {m_si} makes perfect biological sense”, and in preceding sentences. In the SI of AF, algorithm 7 is called: “gated self-attention per row with pair bias” and also in the main article (fig. 3a) it can be seen that the representation of the pair affects the self-attention per row. Maybe you meant something implied and I didn’t understand, if so I’m sorry and kindly apologize.
Anyway, thanks a lot for the article, it’s helping me to penetrate some detail of AF and I really needed a guide!
Thanks for the article, your viewpoint nicely complements what others wrote. Trying to understand AF2, I in fact use a method very similar to the AF2 model itself: Look from many different angles, compose the whole picture from all the bits.
I just spent a few days to get yet another view – better feel for the possible protein geometries. I made a 3D printed molecules kit for this – maybe it can also help some other deeply interested readers coming here. It is at Printables: https://www.printables.com/model/457984-molecules-for-protein-folding